How to Build a Strep A Vaccine
Words by
Eva Amsen

In 1793, Philadelphia physician Benjamin Rush — a signatory of the US Declaration of Independence and close friend of Thomas Jefferson — described a “putrid” sore throat afflicting the city’s population. The disease came on suddenly; a child well at breakfast, Rush claimed, could drop dead by nightfall.
This disease was likely scarlet fever, an ailment caused by Streptococcus pyogenes, from the Greek words streptos (twisted) and pyogenes (pus-forming). Victorians knew the illness as “scarlatina,” Hungarian physician Ignaz Semmelweis, of handwashing fame, named it puerperal fever, and Americans of the 1920s called it “strep throat.”
While there are no global statistics on deaths from scarlet fever, an estimated 10,000 people per year succumbed in England and Wales alone during its peak in the mid-nineteenth century. As antibiotics became widely available in the 1940s, scarlet fever became more manageable and less deadly. Yet, although it may seem a disease of the past, as recently as 2022, a scarlet fever outbreak in the UK infected about sixty thousand people, killing hundreds.

S. pyogenes secretes toxins that disrupt the normal immune response, activating large numbers of T cells and flooding the body with inflammatory molecules. It causes up to one in ten sore throats in adults and one in four in children. Strep throat often inflames the tonsils and can, on occasion, cause ear infections. If left untreated, it occasionally develops into pneumonia, meningitis, or rheumatic fever, which can then lead to long-term rheumatic heart disease, permanently damaging the heart’s valves. Rheumatic heart disease affects 55 million people worldwide, mostly in low- and middle-income countries, and kills about 360,000 people per year, according to the World Health Organization. Meanwhile, Strep A is gradually becoming resistant to common antibiotics, so other means of treatment and prevention are desperately needed.
A vaccine would go a long way toward reducing the spread of Strep A, but development has been impeded by decades of regulatory and scientific challenges. An FDA ban on unpurified Strep A products, driven by safety concerns, meant most funders and researchers stopped working on Strep A vaccines from 1979 until 2006. Even after large-scale efforts resumed, making a vaccine proved tricky; advances in genomics revealed that Strep A has unusually high variation in its surface proteins, meaning that a vaccine against one variant doesn’t necessarily convey immunity against others.
Since the ban was eased, researchers have been making up for lost time, painstakingly mapping the roles of the bacterium’s proteins and carbohydrates. We are closer than ever to a Strep A vaccine, with several candidates now in early-stage clinical trials. The current challenge is to find sufficient funding for the next stages of testing and rollout.
{{signup}}
Vaccination teaches the immune system to recognize disease-causing bacteria or viruses so that it can make antibodies against them if a later infection occurs. Some vaccines contain whole viruses or bacteria, either inactivated or significantly weakened. But these methods have drawbacks; weakened vaccines cannot be given to people with compromised immune systems, because even an attenuated pathogen can cause illness. Also, inactivated vaccines often don’t elicit a strong immune response, thus requiring multiple doses.
Researchers began investigating vaccines that include only antigens — the parts of the pathogen that elicit an immune response — to solve these problems. Antigen vaccines trigger a more specific immune response than an entire (weakened or inactive) pathogen, and also sidestep side effects that might occur from including all parts of the pathogen.
The first step in making an antigen-based Strep A vaccine was to identify its antigens. As these are too small to see with a light microscope, microbiologist Rebecca Lancefield devised a clever series of tests in the 1920s and 1930s to do so. She isolated bacteria from patients with streptococcal infections, cultured the microbes in broth, and then broke open the cells to extract molecules on their surfaces. Then, she injected these molecules into rabbits. After six to eight weeks, the rabbits made antibodies against all of the antigens. Lancefield took this rabbit serum and mixed it with her original bacterial extracts in little glass tubes. If the rabbit serum contained antibodies matching an antigen in that extract, the two would bind, clumping into a cloudy precipitate.
By studying precipitates with extracts taken from many different streptococcal strains from dozens of infected patients, Lancefield figured out that all the streptococci-causing infections shared a unique antigen; a carbohydrate that Lancefield named the “C-substance.”1
Carbohydrates are not the only antigens found on Strep A bacteria, though. These microbes also bristle with helical proteins — called “M protein” — that help the bacterium invade human cells by binding to fibronectin, a protein found on epithelial cells in the throat. M proteins also latch onto antibodies (albeit away from their “recognition” sites), thus helping them evade detection by white blood cells. And Strep A microbes carry other surface proteins, such as SpyCEP, which destroys messenger molecules that would otherwise guide immune cells to the bacteria.
M proteins have received by far the most attention in Strep A vaccine development. One of the researchers working on such a vaccine was Benedict Massell. Between 1965 and 1967, Massell’s team at Harvard Medical School ran a small trial in which they vaccinated 21 healthy children by injecting them with M protein. This trial was seminal because it unintentionally highlighted a safety issue that has shaped Strep A vaccine research to this day.
When Massell published his results in the Journal of the American Medical Association in 1969, he disclosed that two of the 21 children injected with purified M proteins had definitely developed rheumatic fever, while a third had “probable rheumatic fever.” By contrast, Massell found just five cases of rheumatic fever amongst 447 unvaccinated children. His vaccine, then, appeared to increase the odds of rheumatic fever.
Researchers now understand that rheumatic fever and rheumatic heart disease are likely caused by an autoimmune response, due to similarities between parts of the M protein and proteins in the human heart. This wasn’t confirmed until the 1980s, though, so Massell had no idea why his vaccine was causing these serious side effects. He paused the trial, concluding: “Our experience indicates the need for extreme caution in conducting studies with streptococcal vaccine in human subjects.”
Despite Massell’s warnings, researchers at the University of Chicago ran several human challenge studies from 1973 to 1978 to further test M protein vaccines.2 In these trials, volunteers were deliberately infected with a pathogen after vaccination to see whether the vaccine protected them. The Chicago team used smaller doses and a more thoroughly purified M protein than Massell had, believing that the unwanted side effects had originated from other remnants of the bacteria. They also worked with adult volunteers instead of children, providing antibiotics within days to subjects showing signs of illness.
In some challenge trials, the Chicago researchers gave the purified M protein as nasal or throat sprays, which reduced the number of people who got sick after later infections by a factor of two or three, with zero participants showing signs of rheumatic heart disease. Another study with injected vaccines proved even more effective, with only one of 19 vaccinated people becoming ill, versus nine in a similarly sized control group. M protein looked like a promising path toward a vaccine.
But in 1979, the FDA blocked all research on products with “No U.S. Standard of Potency” made from Strep A bacteria. The FDA’s report cited Massell’s findings as evidence for the high risk of heart disease that could occur when using these products. The ban was aimed at Strep A vaccines that used insufficiently thorough preparations of antigens from bacterial cultures; but out of an abundance of caution, many researchers and funders took it as a de facto moratorium on all Strep A vaccine research.
When the FDA finally lifted the ban in 2006, they acknowledged that the regulation had inadvertently discouraged research it had never meant to stop: “Although it was never intended to apply to the development of Group A streptococcal vaccines that had adequate testing, FDA has determined that it has been perceived to cover these products as well, and therefore should be removed in a direct final rule.”
By then, however, research into Strep A vaccines lagged decades behind that of other infectious diseases. For example, the first vaccine against Haemophilus influenzae type b, a leading cause of childhood meningitis, was introduced in 1985, work on the HPV vaccine started in 1995, and polio was eradicated in the Americas by 1994.
Still, it was during this vaccine hiatus that scientists made a discovery which steered the next wave of vaccine candidates. Specifically, in the early 1990s, biologists discovered that the M protein was not a single entity, but rather had many different variants, called “emm types.” There were about 80 known emm types in the early 1990s; today, this figure has risen to over 275. (For comparison, Neisseria meningitidis, the bacterium that causes meningitis, has only 13 known serogroups.)
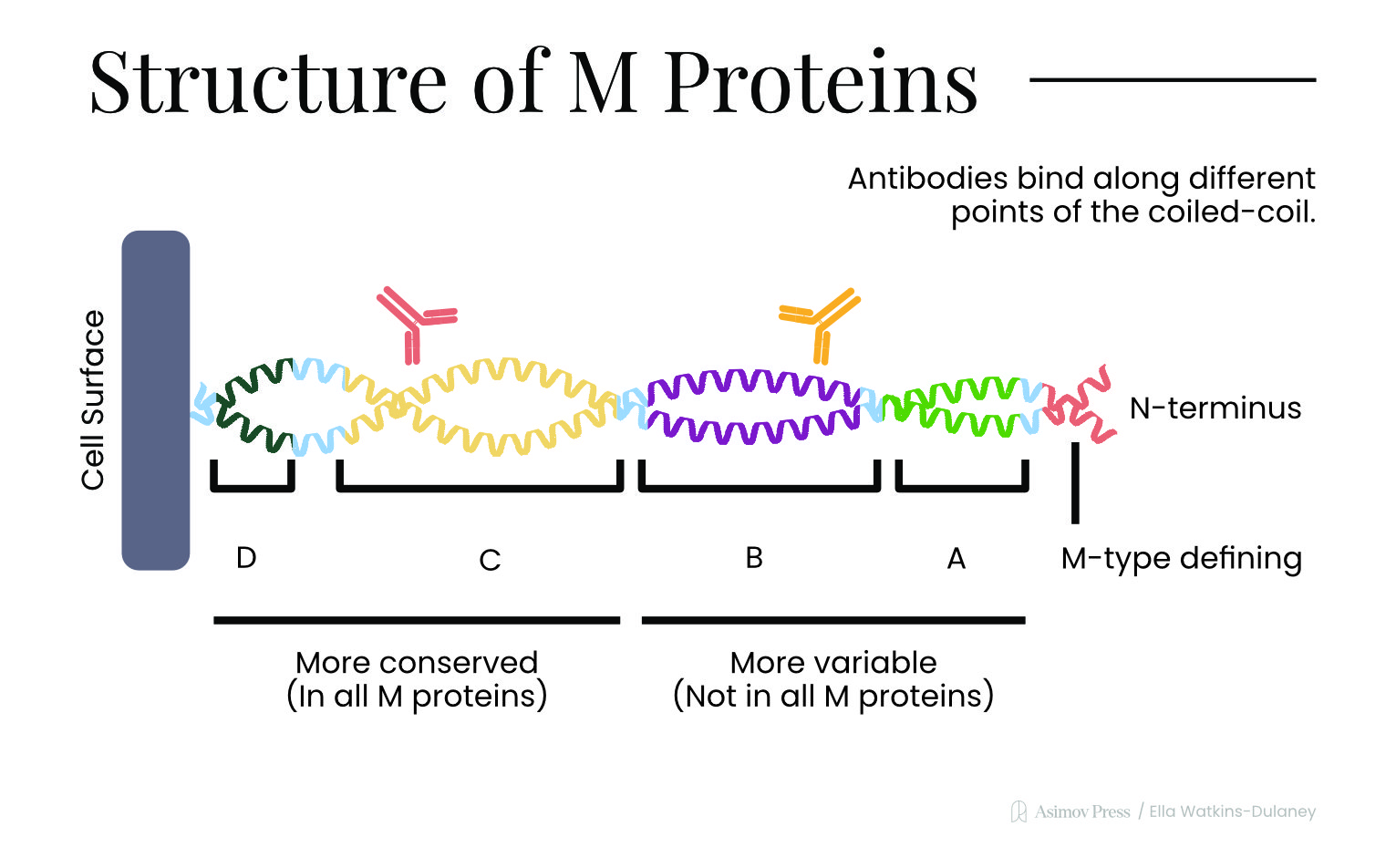
Strep A’s many emm types make it much wilier at avoiding the immune system because antibodies against one version of the M protein do not necessarily recognize others. Over the last 20 years, therefore, researchers have been trying to create Strep A vaccines that can protect against multiple emm types.
“You don’t need to cover 200 emm types to have a significant impact on disease,” says Jim Dale, Professor Emeritus at the University of Tennessee Health Science Center, who spent decades working on Strep A vaccines. “If we could prevent 50 percent of the cases of rheumatic fever, and therefore 50 percent of the cases of rheumatic heart disease, we could have a significant impact on the health of people around the world.” The challenge, then, is figuring out which emm types to target and how to combine them into a single vaccine.
The M protein’s variable bit is actually just a small region near its tip, and Dale has been studying how to use just this “variable region” to make a vaccine since the 1990s. Because his vaccines are built from synthetic peptides designed in the lab — rather than from Strep A bacteria themselves — Dale was able to sidestep the FDA ban entirely.
Since each emm type corresponds to a distinct variant of the M protein, Dale first figured out which variants were most commonly found in Strep A infections or most likely to cause rheumatic fever. His first vaccine design targeted 26 variants based on epidemiological data from North America; an updated version, called StreptAnova, expanded to 30 variants by incorporating data from European infections.3 Those 30 variants together account for the Strep A strains responsible for about 98 percent of strep throat cases in North America, according to Wellcome, and 78 percent of infections in Europe.
To test whether these vaccines could trigger an immune response, Dale’s team injected StreptAnova into rabbits. After a series of immunizations over several weeks, the animals had made antibodies against all 30 M protein variants. These antibodies, when isolated and tested in vitro, killed Strep A bacteria representing 28 of the 30 targeted variants. Surprisingly, the rabbit antibodies also killed 24 of 40 additional Strep A variants that were not included in the vaccine. This suggests that enough similarity exists between different M protein variants for a vaccine to protect beyond its original targets.
Another way to design vaccines which cover many Strep A variants is to avoid the M protein’s variable region altogether, instead targeting its conserved regions. Using this method, researchers in Brazil created a vaccine candidate called StreptInCor, a synthetic peptide made from 55 amino acids taken from that conserved part of the M protein. When injected into mice, it coaxed the animals to make antibodies that recognized and killed many different Strep A strains. The initial vaccine design was published in 2006, with animal results published in 2013.
Whether they target multiple variable regions or one conserved region, these M protein-based vaccines all include only a small part of the original protein to avoid triggering rheumatic heart disease. All these vaccines were designed using only parts of the M protein with no known sequence similarity to any human protein and, just to be sure, lab animals were also tested for autoimmune reactions when immunized.
A third way to circumvent M protein variability is to make vaccines that target Strep A’s characteristic carbohydrate molecules. The problem with this approach, though, is that a robust immune response requires both T and B cells, but carbohydrates on their own tend to activate only B cells, producing a short-lived response. To trigger T cells, the carbohydrate must be coupled to a carrier protein. But since the immune system will also make antibodies against the carrier protein, Helge Dorfmueller at the University of Dundee has been revising these vaccines to use Strep A proteins themselves as carriers. The result is a vaccine with two Strep A antigens — a protein and a carbohydrate — improving the chance of generating antibodies against the bacteria.
So far, the vaccines furthest along in development are all based on a small peptide or a peptide attached to a carbohydrate. But in the last five years, researchers have also begun work on mRNA vaccines against Strep A. Instead of injecting proteins or peptides directly, these vaccines tap into the cell’s own protein-making machinery. When vaccines are delivered as mRNA, the cell uses it as a blueprint to pump out the antigen itself.
This method, developed in the early 2000s by Katalin Karikó and Drew Weissman at the University of Pennsylvania, gained popularity during the COVID-19 pandemic, when Pfizer-BioNTech and Moderna both used it to develop vaccines against SARS-CoV-2. Moderna has since partnered with Dale to work on an mRNA version of a StreptAnova-like vaccine and with researchers at the University of Queensland to develop a vaccine against five other Strep A surface proteins, including SpyCEP.
mRNA vaccines are also cheaper to make, since they are faster to manufacture than protein-based vaccines. For a vaccine producer that already has an mRNA vaccine, switching to a different target can be done in just days (not including testing and quality control) because the process reuses most of the existing workflow. In contrast, it can take weeks or months to adjust a vaccine pipeline to make and purify a new peptide.
With so many Strep A vaccines, many candidates stand ready for clinical testing, but none have advanced to phase 3 trials.
The first to enter human testing was StreptAnova, which completed a phase 1 in 2020. During this trial, StreptAnova was tested in a small group of 23 volunteers, who were monitored for adverse effects or autoimmune reactions. Even after three doses, no volunteers had side effects. One month after receiving the last dose, trial participants also had increased levels of antibodies against 24 of the 30 Strep A variants in their blood, indicating that the vaccine worked. Furthermore, immune cells from their blood after vaccination were able to kill Strep A in cell cultures.
Despite these results, though, StreptAnova has yet to move into the next stages of clinical testing. “The hesitancy of big vaccine manufacturers in taking on the challenge of clinical development of strep A vaccines has been a big roadblock,” says Dale. The previous version of his vaccine, with 26 variable regions, had already passed phase 1 trials with no interest from manufacturers, so Dale hoped that yet another trial of the new, 30-valent vaccine would be enough to convince them. “As it turns out, we were wrong,” he adds. “There was not any greater interest in StreptAnova than there was in the 26-valent vaccine.”
Meanwhile, other vaccines based on the conserved region of the M protein are also in early trials. A conserved-region peptide vaccine completed a phase 1/2 trial in Canada in May 2025, and StreptInCor is currently in a phase 1/2 trial, expected to run until 2028.
The main reason these vaccines have yet to make it into phase 3 trials is funding; vaccine manufacturers are concerned they won’t recoup a sufficient return on their investment. “In general, when a disease or infection affects people in lower-income countries, it gets less attention and less funding,” says Jacob Trefethen, formerly Managing Director of Global Health & Wellbeing at Coefficient Giving.
The Center for Global Development estimates that 82 percent of the vaccine market’s revenue comes from high-income countries that represent only 16 percent of the population. To make vaccines affordable for lower-income countries, Gavi, the Vaccine Alliance, was launched in 2000. Funded by donors including the WHO, UNICEF, World Bank, and the Gates Foundation, Gavi subsidizes vaccine procurement and delivery for countries that can’t afford high prices. For example, vaccines for diphtheria, tetanus, and pertussis — which cost more than $20 per dose in the United States — can be offered to children in low-income countries at less than a dollar per dose through Gavi and Unicef.
This means companies rely on richer countries to add vaccines to their childhood vaccination schedules to compensate for the cost of rolling them out in poorer countries, which tend to have higher disease burdens. That requires regulators and policymakers in wealthy countries to recognize the long-term global impact and value of such vaccines, which is an easier case to make when more trials are funded.
The investment seems well worth it. Coefficient Giving has researched how much funding different infectious diseases had available, and compared this to the number of people affected and the number of vaccines in development. “In our nine years of funding vaccine R&D, Strep A jumps out as one of the most important, neglected, and tractable pathogens to work on,” says Trefethen. Strep A’s disease burden is expressed as a disability-adjusted life year (DALY) of 17.4 million, not far from the DALY of 18.4 million for HIV/AIDS. But the estimated annual funding available to both is staggeringly different: $1.5 billion for HIV/AIDS versus $14 million for Strep A.
While a lack of investment affects vaccine development for many infectious diseases, Strep A faces additional hurdles. After the FDA ban was lifted, researchers have been extra vigilant about designing vaccines that are less likely to trigger rheumatic fever. However, the ban still serves as a reminder that any Strep A vaccine they develop needs additional safety checks beyond those required for vaccines against other infectious diseases. Even though relatively easy ways to implement this exist, for example, by using echocardiograms to test for heart damage among trial participants, manufacturers have been hesitant to invest the extra time and cost.
The final challenge is the lag time in determining whether a vaccine really protects against rheumatic heart disease. The condition develops over many years, so people may not show symptoms until decades after their first Strep A infection. That makes it difficult for manufacturers to demonstrate that their vaccines work. “There needs to be a better link with the endpoints that a company will use,” says Jerome Kim, Director General of the International Vaccine Institute (IVI) and Co-Chair of the Strep A Vaccine Global Consortium (SAVAC). Here, too, a solution might already exist. In 2024, cardiologist Andrea Beaton showed that early heart lesions linked to Strep A infections are a reliable predictor of rheumatic heart disease later in life, which means vaccine manufacturers wouldn’t have to wait decades to find out if a vaccine has worked.
Another part of the solution is simply gathering more data. “I think Big Pharma is waiting for vaccine developers, for academics like me, to show them a clear winner,” says Dale. That could mean running additional, smaller trials to provide further confidence that a larger one is worth funding.
Human challenge trials may help. In 2021, Andrew Steer and Josh Osowicki of Murdoch Children’s Research Institute in Australia designed new “controlled human infection models” that use lower, safer doses of Strep A than those from the 1970s. “New human challenge models should allow for quicker testing of different options,” says Trefethen, because participants are deliberately infected with Strep A, so studies can move faster than trials where infection must occur naturally. None of the vaccine candidates currently awaiting funds for phase 3 trials have yet gone through human challenge trials. Thus, their initiation could provide much-needed additional data to support these vaccines.
All new trial and safety data will also need to be communicated to organizations responsible for the approval and global rollout of new vaccines, such as the WHO and health departments worldwide. SAVAC and the Australian Strep A Vaccine Initiative have taken on this coordination. Kim explains that the WHO’s Strategic Advisory Group of Experts on Immunization can recommend a vaccine based on trial data, while SAVAC reaches out to Gavi to ensure they can make it available in low- and middle-income countries.
“We have to convince everybody that it’s something worth moving forward on because the burden is high, the technology is not so daunting, and the risks are reduced by all the work that we’ve done,” says Kim.
With that said, the technology has improved, and the risks have dropped precipitously over the last 20 years. “My real hope is that the problem may be mostly related to attention and funding,” says Trefethen, “those are easier problems to solve than scientific ones.” If Trefethen is right, the prognosis looks good. With vaccine candidates now in clinical trials, a better understanding of how Strep A infects and evades our immune system, and improved ways to monitor and predict rheumatic heart disease, the only hurdles remaining are tractable ones (attention and funding) rather than ignorance of the science behind this complex and wily disease.
{{divider}}
Eva Amsen is a science writer in London with a PhD in biochemistry. She has written about biology, chemistry, environmental sciences, and the overlap of science and the arts for various publications, including Undark, The Observer (Guardian), Nature, Hakai, and Nautilus. You can find more on her website: evaamsen.com
Cite: Amsen, E. "How to Make a Strep A Vaccine." Asimov Press (2026). DOI: 10.62211/29he-52tw