Making Cells Young
Words by
José Luis Ricón
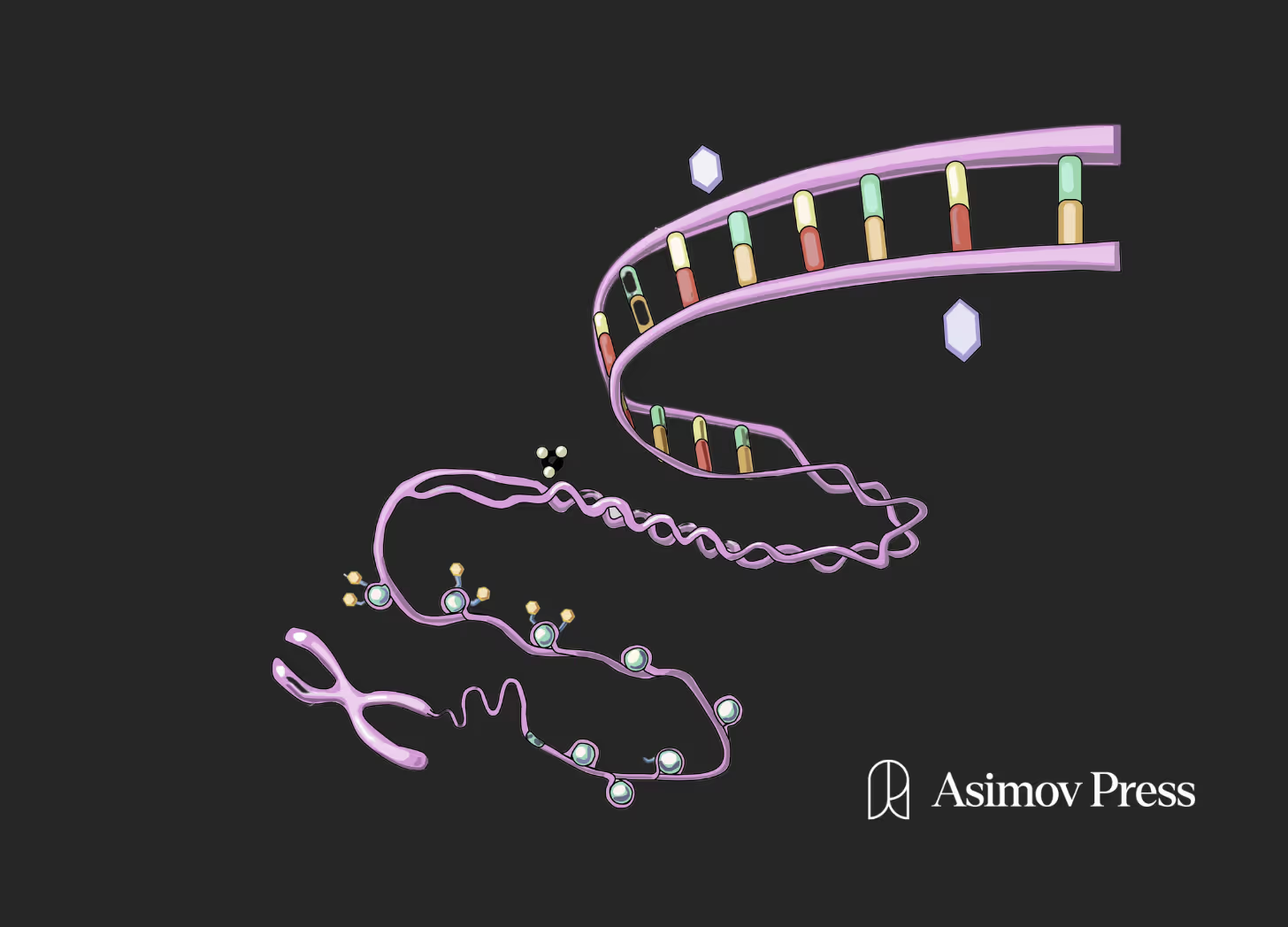
Not everyone ages in the same way. Some people go deaf and others go blind. Most people die from cardiovascular disease, while others succumb to Alzheimer’s and cancer. Some people live in relatively good health until the very end, whereas others suffer a slow but relentless decline. Regardless of how quickly this process transpires, aging tends to involve a cascade of bodily deterioration that is challenging to treat holistically.
Take cardiovascular disease as an example. The most common approach to treatment is to attempt to reduce some causal biomarkers, such as cholesterol, with drugs like statins. However, this approach is imperfect because it only addresses one manifestation of the disease, rather than its root. As a person ages, blood pressure also increases. Additional drugs are then typically introduced to lower it again. So it proceeds—as new aging symptoms appear, they are treated with yet another drug as in a game of whack-a-mole.
And that’s just one example. The aging brain similarly deteriorates over time. Microglia, the “garbage collectors” of the brain that eat up dead cells and protein aggregates, slowly lose their functionality. The brain becomes inflamed, and neurons begin to die. The glymphatic system, tasked with cleaning out the brain, also becomes less efficient. The blood-brain barrier gets leakier and begins to let in biomolecules that should be kept out.
A similar deterioration occurs in fibroblasts in the skin, hematopoietic stem cells in the bone marrow, and hepatocytes in the liver. Aging wreaks havoc regardless of a cell’s position in the body. All struggle to perform functions they could previously do with ease. Old cells have fewer functional mitochondria, altered expression of genes, and decreased ability to recycle damaged proteins. But why?
The field of aging has been debating this question for decades. Peter Medawar, a British-Brazilian biologist who is often called the “father of organ transplantation,” referred to aging in 1951 as “an unsolved problem of biology.” In the decades since, many others have tried to find causes (or, more ambitiously, “the” cause) of aging, putting forth various theories that fluctuated in both popularity and credibility. It is against this backdrop that a new explanation for the phenomenon of aging, called the “information theory,” presents itself.
The theory states that, at least at the cellular level, aging is a phenomenon that is mainly driven by the loss of epigenetic information. It also posits that, by reverting cells to their pluripotent state, we can effectively reprogram them into once again behaving like young cells. It is the hope that, one day soon, this technique will allow us to replace aged cells with revitalized ones, fighting back against cellular aging.
{{signup}}
Most cells (excluding T-cells and B cells after V(D)J recombination) in our body contain the same DNA but appear and behave in distinct ways: A neuron looks and acts very differently from a hepatocyte. This is due to epigenetics, defined here as the relatively stable set of modifications to DNA, like CpG methylation, that determine how DNA is interpreted in a given cell.
While DNA is relatively stable, the epigenome is not; it has to orchestrate changes in cell state, cell type, and more. As a result of this instability, we lose epigenetic information in our cells as we age. Our cells forget what they are supposed to be doing and when: which genes should be expressed in each situation. This “information” is also the basis of the now-popular “epigenetic clocks,” tools that quantify the number of epigenetic changes carried on the genome to estimate the biological age of a human, a fish, or any other organism.
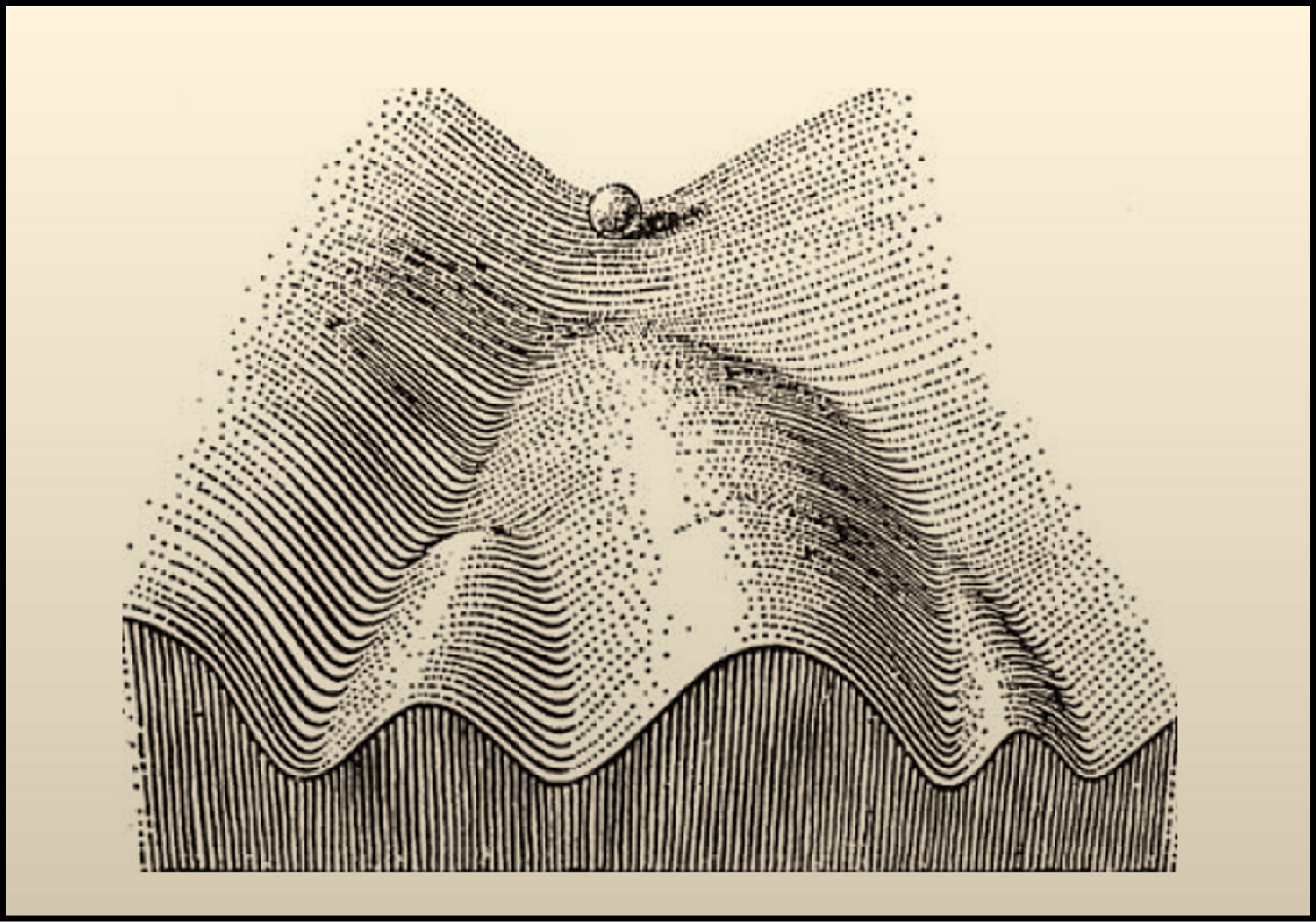
While it may seem presumptuous to say that aging has a single major driver even at the single-cell level, this is not the first time the field has been presented with a seemingly definitive theory of the phenomenon it studies.
In 1961, a medical microbiologist at Stanford, named Leonard Hayflick, noticed that cells stop growing after a while when they are cultured in a dish. Not only that, but the older the cells were to begin with, the fewer times they would divide, which made it seem as if the cells had some sort of built-in clock.
Other scientists, such as Alexey Olovnikov, Elizabeth Blackburn, and Carol Greider, further investigated why this is. They discovered that chromosomes have so-called telomeres at their ends, which are repetitive sections of DNA that shorten every time a cell divides. If a telomere becomes too short, the cells stop dividing. They also discovered that some cells express an enzyme, called telomerase, that can help preserve telomere length. When the gene encoding telomerase is removed or mutated in Tetrahymena, a single-celled eukaryote, the cells grow slower and then die after a few weeks. These experiments, and others, suggested that telomeres could be a driver of aging.
The Geron Corporation was formed in 1990 to pursue therapeutics based on telomere biology, and its public relations department worked hard to inculcate this idea. It worked to spectacular effect. To this day, people who work in the aging field are often asked if “that’s the telomeres thing.” And the answer is: “Not quite.”
As a whole, those who worked in the field never really believed that telomeres were the driver of aging, although they generally acknowledged that their shortening could be a contributor, with variable importance across species. In humans, for example, telomeres don’t seem to matter as much as in other organisms. Our telomeres shorten at a very slow rate. Lengthening the telomeres of a cell does allow it to divide more times, but it doesn’t reverse other hallmarks of aging, such as epigenetic damage, at the cellular level.
In contrast, evidence for epigenetic-driven aging is much stronger than any hypotheses that have come before, including telomere-driven aging. The first piece of evidence that epigenetic damage is a key driver of aging goes all the way back to work by a British biologist, named John Gurdon.
In 1975, Gurdon took the nucleus from a somatic cell from an old frog and placed it into an enucleated oocyte: basically an egg without a nucleus. In some cases, the re-nucleated oocyte gave rise to a healthy animal. And even though the nucleus—and all the DNA inside of it—came from an old frog, the resulting frogs were young and healthy. Today, we know that cloned animals made using somatic cell nuclear transfer do not age prematurely. Rather, they live normal lifespans and remain as healthy as a non-cloned member of their species, supporting the idea that the process of embryogenesis can reset whatever damage the nucleus might have experienced during the aging process, except DNA mutations that do get passed on to the next generation.
Decades later, Gurdon’s ideas were harnessed by a Japanese scientist, named Shinya Yamanaka, who figured out how to make pluripotent stem cells (iPSC)—cells that can give rise to any other cell in the body—by “reprogramming” normal, somatic cells using just four proteins: Oct4, Sox2, Klf4, and c-Myc. Both Yamanaka and Gurdon shared the 2012 Nobel Prize in Physiology or Medicine.

When a somatic cell is reprogrammed into an iPSC, either by using Yamanaka’s method or small molecules that mimic the four proteins, the epigenomes of the reprogrammed cells are effectively “wiped clean” and reverted into a state comparable to that of an embryonic stem cell.
Interestingly, if one makes a cell type out of another cell that is not an iPSC (a process called transdifferentiation or direct reprogramming), the resulting cell is not de-aged. There’s something special to the generation of these embryonic stem cell-like cells. After all, the molecular pathways involved in total reprogramming evolved to allow aged animals to give birth to non-aged babies so that an age-resetting effect happens with each generation.
So far the only method that has proven to completely reverse cellular aging is transiting through the iPSC/embryonic state. Experiments have demonstrated that it’s possible to examine old cells before iPSC conversion and then compare them to the resulting cells obtained from the iPSC.
In 2011, scientists in Montpellier, France took fibroblast cells and reprogrammed them into a pluripotent state. They looked at telomere length, mitochondrial function, gene expression, and oxidative stress. They ultimately concluded that the resulting cells had “been completely rid of their former aged cellular phenotype.” And others, more recently, have observed the same thing. In 2015, a review from scientists at the Memorial Sloan Kettering Cancer Center, in a more comprehensive analysis, similarly concluded: “Remarkably, every single age-related parameter expressed in the aged donor fibroblast population was reset.” This finding has been replicated in other cell types, including neurons. The pluripotent stem cells, made via cellular reprogramming, are functionally equivalent regardless of whether they are made from old cells or young cells.
One wrinkle in this is that cellular reprogramming does not undo DNA mutations, which can cause cancer and other diseases. Still, everything else that we have been able to measure so far in reprogrammed cells, including chromatin accessibility, gene expression, mitochondrial function, or senescence markers, are rejuvenated after reprogramming and resemble a young, healthy cell, despite the mutated genome sequence. With this caveat about its limitations, then, we can say that aging is effectively solved in vitro, given available published evidence.
Now the question is: Can cellular reprogramming similarly address epigenome damage as a root cause of aging at the organism level? Perhaps, but it won’t be simple.
Although much of the experimental evidence described thus far is about individual cells grown in a laboratory, it’s not unreasonable to extrapolate from the existing evidence and infer that the aging of the cell must have something to do with the aging of the organism as a whole. If we can make the cells in an organism younger, surely we will have taken one step closer to true age reversal. Many people, including some wealthy investors, have bought into this reasoning, and with it, the larger ambition implicit in “solving” this problem of biology—discovering a fountain of youth. This has led to the largest wave of funding rounds in recent history for any company working on aging: Altos Labs ($3B), Retro Biosciences ($180M), and NewLimit ($150M).
If this epigenetic-driven theory of aging is roughly correct, reprogramming will solve a lot: Some cells can be easily removed from the body, reprogrammed in carefully controlled laboratory conditions (such as hematopoietic stem cells or T-cells), and then re-infused into the body via established methods, such as bone marrow transplantation. While this could help revitalize our immune system or replenish certain stem cells, it still leaves us with our organs, which won’t be so easy to reprogram.
This is where another technique, called partial reprogramming, comes in. If full reprogramming is fully de-aging a cell (but making it lose its identity along the way), partial reprogramming leverages the same biology to push the cell towards an iPSC state, without getting it there entirely. Instead, partial reprogramming reverts a cell to a roughly “halfway” point, whereby the cell can return to its type of origin, without losing identity, while being rejuvenated in the process.
Partial reprogramming is typically done either by dosing the transcription factors used for reprogramming into cells or an organ intermittently, or by coaxing cells to express these transcription factors using an inducible system, such as Tet-On. This is an approach that lets the cells retain their identity while gaining some of the rejuvenation effects of the iPSC conversion process. Though partial reprogramming is a new field, scientists have already been able to extend the lifespan of wildtype and progeroid mice, as well as improve their health in various ways: metabolic function as measured by a glucose tolerance test, cognitive function, or vision loss.
But there’s a catch: unlike with the small molecule drugs we are used to, the side effects of reprogramming can be far worse than a headache or nausea. If a few cells accidentally get reprogrammed all the way to an iPSC state, cancer can result. The iPSC cells can form teratomas, a type of tumor that originates from these pluripotent cells, which can expand and differentiate in a chaotic fashion.
This is why it is crucial to control reprogramming in every single cell that gets reprogrammed. We can’t do this just by varying the dose of the therapy; some cells might still get too much by chance and form teratomas as a result.
Clearly, we will need better tools to precisely regulate the expression of transcription factors to guide cellular reprogramming. Historically, the tools we have had to manipulate biology have been binary: completely knocking out a gene (0) or turning on a different gene (1), but rarely regulating how much of a gene or when a gene should be expressed. But cells don’t live their life this way.
Rather, they rely on exquisite pathways that regulate gene expression depending on cell state and environmental cues—something we could also call a “gene circuit.” For reprogramming, we may want to turn on the process but then shut it down permanently once the process has progressed “enough,” with “enough” defined in terms of the output of a gene circuit, thus using biology’s own structures for controlling genetic programs.
We don’t yet know the full scope of what reprogramming can do in vivo. Tissues change in ways that do not merely involve cellular aging. The kinds of cells present and the structures they form (like glomeruli in the kidney or myofiber types in the muscle) are altered as well. Can cellular rejuvenation alone remodel tissues back to what they look like in young individuals? If the answer is “no,” then interventions beyond reprogramming will be required for comprehensive rejuvenation. Perhaps some organs can only truly be de-aged by replacing them completely, as was proposed by Albert Einstein College of Medicine scientist Jean Hébert in Replacing Aging.
As of today, we don’t know if reprogramming can fix problems in a tissue beyond those in the cells that get reprogrammed. Some parts of aging preclude reprogramming in the first place. Imagine someone who has lost their teeth. There are no teeth left to de-age anymore, and the teeth don’t grow back a third time naturally. Reversing that specific aspect of age-related decline will likely need a very specialized intervention that goes beyond the kind of reprogramming I have described.
We have yet to fully explore and stress test what reprogramming in its multiple variants can do to address aging. However, as research advances, we will better understand what reprogramming can and can’t do in the context of safe and efficacious therapies applied either in vivo or ex vivo. It’s unlikely that it will be the only thing we will ever need to tackle human aging, but progress so far gives us grounds to be hopeful. With further research, we may, at long last, make a sizable dent in the root cause of most human suffering—aging and its associated devastations.
{{divider}}
José Luis Ricón blogs at nintil.com and is Head of Theory at Retro Biosciences.
Cite this essay: José Luis Ricón. “Making Cells Young.” Asimov Press (2024). DOI: https://doi.org/10.62211/63ru-88pp
This article was published on March 12, 2024.