A Brief History of Bioinformatics Software
Words by
Ella Watkins-Dulaney

When protein sequencing was invented in the late 1950s, biologists found themselves faced with the enormous task of managing and analyzing long strings of apparently random amino acids. The difficulty was that most humans can only remember a string of random values around seven items long, which is 67 times shorter than the average protein sequence. Fortunately, scientists’ growing need for sequencing coincided with the development of computers, which helped to make sense of the overwhelming influx of data.
Although the term “bioinformatics” was coined in 1970 by Dutch theoretical biologists Paulien Hogeweg and Ben Hesper, the first bona fide bioinformatician was a quantum chemist, Margaret Belle (née Oakley) Dayhoff. Born in 1925 in Philadelphia to Ruth Clark, a high school math teacher, and Kenneth W. Oakley, a small business owner, Margaret was academically gifted and flourished in the sciences, a notable achievement for a woman in her day.
Dayhoff completed her PhD at Columbia University in just three years, receiving her diploma in 1948. She was one of the first to use computers to solve quantum chemistry problems, a process she described in her thesis, “Punched Card Calculation of Resonance Energies.”1 In the decade or so that followed, Dayhoff focused on raising her children, interrupting this professional hiatus only briefly to take a postdoctoral position in computational chemistry from 1957-1959 at the University of Maryland.
When she tried to resume full-time work in 1960, Dayhoff was surprised to have her funding application rejected by the NIH, which cited her “time off.” This led her to work with an acquaintance of her husband, Robert Ledley, a biophysicist who had just established the National Biomedical Research Foundation (NBRF), an organization in Silver Spring, Maryland dedicated to promoting the use of computers in biomedical research.


Ledley was an early believer in “computerizing” biology and medicine. He explained his motivation as a hunch that computers would one day be “analogous to the staff of a laboratory” and that each program would have “a function to perform just as a laboratory has people with each a job to perform: cleaning people, technicians, senior research workers, a librarian, a machinist, etc. The programmer and the protein chemist have been upgraded to the chief of the computer staff.” Though we take this idea for granted, it was unusual in Ledley’s day. Many biologists at the time rejected computers, and some were outright hostile. Dayhoff later recalled one biochemist who refused to share data or collaborate, stating “I am not a theorizer.”
Still, Ledley and Dayhoff pursued this opportunity to merge biology and computation. Combining their expertise, the two endeavored to create a computer program that could assemble full protein sequences based on results from the Edman Degradation reaction, a sequencing technique developed in 1950. The Edman Degradation reaction was limited in that it could only be used to sequence strings of about 60 amino acids before the reaction stalled out. To handle longer proteins, these shorter strings were broken up and sequenced as a set of overlapping peptide fragments. Assembling the full sequence required a researcher to compare the fragments one by one to piece them together.
{{signup}}
Dayhoff’s program was published in 1962. Named COMPROTEIN, it freed scientists from the drudgery of protein alignment. When tested against ribonuclease, a sequence that had taken scientists months to solve manually, the program correctly arrived at the sequence in a matter of minutes.


The ability to automate Edman Degradation reactions with programs like COMPROTEIN led to a rapid leap in the number of sequenced proteins. In 1965, Dayhoff, Richard Eck, and colleagues published the Atlas of Protein Sequence and Structure, the first ever biological sequence database. Notably, the Atlas also contained the first published use of the single-letter amino acid abbreviations (Tryptophan: Y, Glycine: G, Lysine: K, etc.), still employed today.2 By the end of the decade, the collection contained about 1,000 full protein sequences.

These protein sequences allowed scientists to interrogate evolution and phylogeny like never before. While building phylogenetic trees, a branching graph that organizes creatures by how closely related they are, dates back to Charles Darwin himself, access to molecular data and computation allowed scientists to compare organisms based on sequence data rather than observation alone. The Atlas data led to the fundamental realization that sequence similarity was proportional to evolutionary relatedness: as organisms accrue mutations, they separate into distinct species. Closely related organisms will share more protein sequences, having had less time to drift apart from one another genetically.
The first sequence-based phylogenetic trees relied upon closely related sequences that were easy to align and compare visually. In 1966, Dayhoff and Eck published one of the first computer-deduced phylogenies from molecular sequences for a highly conserved iron-sulfur protein called ferredoxin. Dayhoff presented the phylogenetic tree to the public in Scientific American in 1969, writing, “Each protein sequence that is established, each evolutionary mechanism that is illuminated, each major innovation in phylogenetic history that is revealed will improve our understanding of the history of life.”
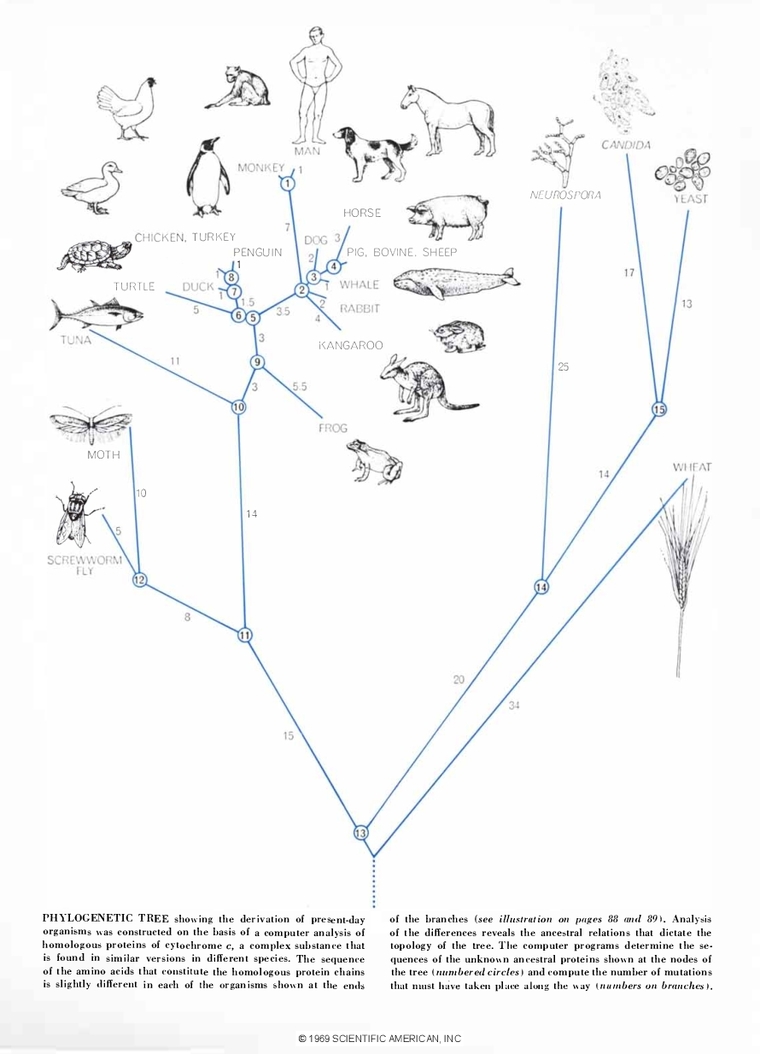
However, this strategy quickly proved insufficient for more distantly related sequences. This pushed scientists to develop multiple sequence alignment (MSA) algorithms that could compare three or more sequences and handle differences in sequence length arising from mutations like insertions or deletions. Their challenge was to create functional algorithms that were memory and time efficient for the hardware of the era. After alignment, they also needed to find a way to calculate sequence similarity to reflect how these mutations correlated to evolutionary relatedness.
In 1970, Saul Needleman and Christian Wunsch presented a dynamic programming approach that could align two sequences of different lengths. They represented these differences by using gaps to shift the frame of each sequence to accommodate insertions or deletions and maximize the number of amino acids that matched one another. However, their algorithm was computationally too slow to apply to multiple sequences.3

Dayhoff and Eck continued to work on this challenge. In 1978, they published a mathematical framework for calculating sequence similarity after the completion of alignments, a method conceptually similar to the algorithms used today. This “similarity score” was more biologically relevant than previous attempts, capturing evolutionary distance by the number of mutations between two sequences. And finally, in 1987, Da-Fei Feng and Russell F. Doolittle published the first truly practical approach to MSA. They used a “progressive sequence alignment” that initially performed a Needleman-Wunsch alignment for all sequence pairs, then extracted similarity scores for each alignment and built a phylogenetic tree based on the comparison of those scores.4
Even during the early days of bioinformatics, Dayhoff and contemporaries realized that these programs could one day be applied to genes once DNA sequencing was developed. Ironically, the man who would finally unlock ribonucleic acid sequencing, Frederick Sanger, was self-admittedly “rather reluctant” to adopt computers. For Sanger, sequence data was an enjoyable puzzle, and employing computers might “take [away] some of the pleasure” that he got from “looking through the sequences and seeing what could be made of them.”

By the 1970s, though, Sanger had accepted that computation was necessary to handle increasingly lengthy sequences. As his group at the Laboratory of Molecular Biology in Cambridge pursued the sequence of the ΦX174 virus, data was split among nine different researchers’ lab notebooks. A British-Canadian biochemist, Michael Smith, was tasked with collecting and organizing these sequences. Since most of Sanger’s group lacked computational experience, Smith recruited his brother-in-law, Duncan McCallum, who routinely used computers to process administrative data in the management division of the chemical multinational Ciba-Geigy. The pair wrote the first programs to analyze DNA sequence data. Their programs were also used to compile the first full DNA genome sequence, ΦX174, for Sanger’s seminal 1977 publication, considered the beginning of the DNA sequencing era.

When Smith returned to British Columbia, Sanger turned to LMB colleague Rodger Staden to help continue the computational work. Staden, a mathematical physicist by training, expanded and adapted McCallum and Smith’s original programs into the “Staden Package,” able to run on minicomputers rather than mainframes. Staden and colleagues continued to develop this package until 2005, and their software is still available to download.
Our DNA and protein sequence databases grew alongside our ability to manipulate genes with techniques like the polymerase chain reaction and restriction enzyme cloning. As these developments required easier ways to share sequences and manage cloning experiments, it was fortunate that the internet emerged at just this time, in 1969. The first wave of ready-to-use microcomputers followed less than a decade later, released to the public in 1977.
Biologists were among the first to use the internet to share information. Soon, several online databases were established, including the Protein Data Bank (PDB) in 1971, and the Protein Information Resource (PIR) in 1984, led by none other than Dayhoff herself at the NBRF. (Regrettably, she passed away shortly before the project’s completion.)
Also in 1984, the University of Wisconsin Genetics Computer Group published the eponymous “GCG” or “Wisconsin Package,” which initially contained 33 command-line tools for the manipulation of DNA, RNA, and protein sequences. While the Staden Package had focused on DNA assembly, the Wisconsin Package included programs for a variety of tasks, like sequence alignment, identifying protein-coding regions in DNA (called “open reading frames,” or ORFs), translating DNA to the corresponding protein sequence, and finding restriction enzyme cut sites for cloning.
Bioinformatics software continued to evolve steadily over the next 40 years, though this stage was mostly defined by improvements in speed, scale, and user experience. Commercial entities, like Geneious, SnapGene, and Benchling, began to offer programs that dramatically reduced the computer expertise needed for simple bioinformatics tasks like sequence alignment and DNA manipulation, core skills for the modern molecular biologist. However, the field has also welcomed numerous public and open-source programs, such as BLAST and BioPython.
It is hard to believe that in under 70 years we have gone from a handful of painstakingly gathered protein sequences to databases like Genbank, RefSeq, UniProt, and PDB that boast billions of unique sequences. These structural datasets, gathered by hundreds of scientists over decades, were crucial for the development of AlphaFold, an AI model for predicting 3D structure from amino acid sequences that won the 2024 Nobel Prize in Chemistry. Much as the Atlas helped to unlock a deeper understanding of phylogeny and evolution in 1965, these sequences are ushering in a new era of bioinformatics driven by machine learning.
{{divider}}
Ella Watkins-Dulaney holds a PhD in bioengineering from the California Institute of Technology. She is now a sci-comm freelancer and the Art Director for Asimov Press.
Cite: Watkins-Dulaney, E. “A Brief History of Bioinformatics Software.” Asimov Press (2026). DOI: 10.62211/72hw-48jh